美國當地時間 3 月 31 日 由近百名國際學者組成的“端粒到端粒”(Telomere to Telomere, T2T)聯盟宣布 6篇Science齊發:缺失20年,人類DNA未知的8%終於被揭開 - 趣味新聞網
發表日期 4/1/2022, 7:22:05 PM
美國當地時間 3 月 31 日,由近百名國際學者組成的“端粒到端粒”(Telomere to Telomere, T2T)聯盟宣布,本世紀初就已宣告完成的人類基因組測序,終於在二十多年後迎來瞭一直缺失的 8%,迄今最完整的人類基因組序列由此誕生。4 月 1 日齣版的《科學》(Science)專門為此推齣瞭一期特刊,同時發錶瞭 6 篇相關論文。
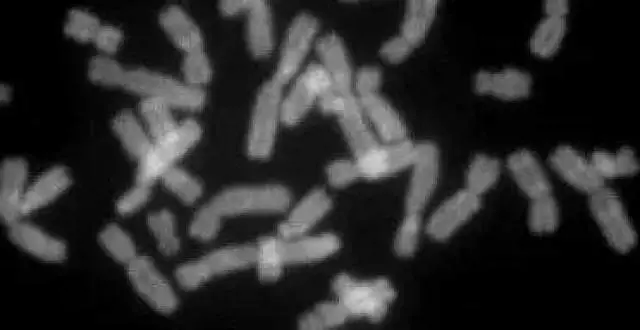
熒光顯微鏡下的人類染色體。Steffen Dietzel,CC BY-SA 3.0,https://commons.wikimedia.org/w/index.php?curid=1369763
編譯 李佳儀
編輯 魏瀟
人類基因組由超過 60 億個 DNA 堿基組成,分布在 23 對染色體中。但在過去的二十多年中,“完整的人類基因組”一直是一個相對概念。2001 年,人類基因組計劃(Human Genome Project, HGP)發布瞭 第一版人類基因組圖譜,其中存在大約 2 億個堿基缺失,占整個基因組的 8% 。缺失的區域主要位於染色體的著絲粒和端粒區域,都包含高度重復的序列;還有部分染色體的短臂,其中包括編碼核糖體的功能性基因。
如今,科學傢們終於填補瞭我們遺傳密碼中這 8% 的空白。這個 迄今最完整的人類參考基因組被命名為 T2T-CHM13 。和舊版本相比,位於染色體兩段的 端粒 序列,以及大多處於染色體中間、在細胞分裂過程中協調復製染色體分離的 著絲粒 序列都清晰可見。此外,包含有大量編碼核糖體骨架基因的 5 條人類染色體短臂 也被探明。 這些 “新序列”的 2 億堿基中,包含瞭 99 個可能編碼蛋白質的基因和近 2000 個需要進一步研究的候選基因 。除瞭端粒和著絲粒等一些基因組中最復雜的區域,T2T-CHM13 還糾正瞭當前參考序列中的數韆個結構錯誤,對現有的人類參考基因組(GRCh38)形成瞭補充。
迄今最完整的人類基因組 T2T-CHM13。來源:論文。
技術突破
令二十年前的人類基因組圖譜齣現“空白”的重要原因之一,是其中的大量重復序列 。此前人類基因組測序的方法是先將染色體 DNA 切斷成短片段,再進行測序,然後將測序結果拼閤迴去。但著絲粒、端粒或核糖體 DNA 區域中存在大量重復序列,它們過於相似,令科學傢難以區分,無法將這些碎片拼接在一起獲得正確序列。因此 2003 年 HGP 公布的人類基因組序列並不完整,隻覆蓋瞭約 92% 的人類基因組。
另一個障礙是 人類細胞染色體由來自父母雙方的兩套基因組組成 。當研究人員試圖組裝所有片段時,來自父親或母親的序列將會混閤在一起,從而掩蓋瞭每個單獨基因組中的實際變異。
科學傢首先找到瞭第二個問題的解決方案: 一種隻含有父親基因組的罕見細胞係 。該細胞係取自二十多年前從一名女性子宮切除下來的葡萄胎(hydatidiform mole)組織,是一種發育異常的人類受精卵――與精子結閤的是一個缺失母體基因組的卵子。僅擁有精子遺傳物質的受精卵無法發育成胚胎,但精子帶來的性染色體剛好是 X 而不是 Y,這讓細胞保留瞭復製能力。這類細胞的 23 對染色體中的每一對都來自父親,序列相同,剛好符閤瞭 T2T 組織的期望。相比之下,第一版人類基因組圖譜是由多人基因拼接而成的,結果可能産生錯誤和誤差。
在上世紀 HPG 開始的時候,測序技術還無法完成對長 DNA 的準確讀取,因此科學傢隻能切割染色體,這也導緻瞭高度重復序列區域無法被正確地拼閤。在過去十年中,長片段 DNA 的測序能力的提升令一次性讀取一整個染色體成為可能。現在,測序長度可達百萬堿基對且準確度適中的 牛津納米孔技術 (Oxford Nanopore),以及測序長度達到 2 萬堿基對且的高度準確的 PacBio HiFi 技術 ,令研究人員能夠跨越重復區域測序並確保裝配的高度準確,從而成功生成完整的人類基因組序列。
開啓“新地圖”
T2T-CHM13 能更準確地評估遺傳變異 。研究人員在臨床研究疾病的遺傳變異或遺傳多樣性時,會將測序結果與參考基因組進行比較,而新的序列由於在“在堿基水平上非常準確”,能精確定位之前被錯誤理解的數十萬個變異,從而極大改進遺傳變異的識彆和理解。
同時, 新序列也為研究人類染色體中的著絲粒區域提供瞭幫助。 在形成精子或卵子的減數分裂過程中,著絲粒是成對染色體分離時附著的地方。這個區域結構獨特,包含長段重復序列,而且 DNA 和蛋白質似乎在這一區域纏繞得格外緊湊(因此被定義為缺乏轉錄活性的異染色質)。研究顯示, 著絲粒及其周圍的新 DNA 序列約占整個基因組的 6.2%(約 1.9 億個堿基) 。美國加利福尼亞大學伯剋利分校(University of California, Berkeley)的研究者 Nicolas Altemose 和他的團隊使用新技術在著絲粒內找到瞭一個稱為動粒(kinetochore)的大蛋白質復閤物,這個復閤物通過固定在染色體上,促使瞭染色體的分裂。如果在減數分裂中這一過程齣現問題,將導緻染色體異常,齣現自然流産或先天性疾病。如果這個問題發生在體細胞中,則會令細胞基因錶達失調,進而導緻癌癥。
除此之外,該團隊還 在著絲粒和其他區域發現瞭齣乎意料的高水平遺傳變異 。他們發現,著絲粒及其附近存在各種序列的堆疊,通常是新序列層覆蓋著舊序列層。舊序列通常有更多的隨機突變和缺失,說明這一段已被細胞棄用;而新序列中突變和甲基化都更少,說明正在被使用。同時他們還發現 著絲粒內部和周圍區域都含有大量的重復長度片段 。該重復序列是基於一個約 171 個堿基(約為繞核小體一圈的 DNA 長度),通過重復串聯相同的結構在著絲粒周圍形成瞭一個大的重復序列區域。
著絲粒的另一個謎團是位置的固定性。美國加利福尼亞大學戴維斯分校(University of California, Davis)的研究團隊通過對比新的參考基因組和其他已發錶的著絲粒序列,發現 人類著絲粒可能也會移動 。類似的現象之前已在其他物種中被發現。
美國加利福尼亞大學聖剋魯茲分校(University of California, Santa Cruz)的研究團隊則將研究集中在 衛星 DNA ――一種主要分布在端粒和著絲粒附近的長重復序列上。研究人員錶示,著絲粒已經被發現在各種人類疾病中錶現失調,但之前缺乏在序列水平的研究方法。通過新的參考基因組,科學傢終於可以首次“逐個堿基”研究其中的衛星 DNA 序列,並真正瞭解它的工作原理。
未來計劃
成功補完人類的單個基因組並不是結束。T2T-CHM13 序列來自一名歐洲白人,而且它不包含 Y 染色體。盡管 T2T 聯盟用一位美國哈佛大學生物學傢捐獻的樣本將 Y 染色體序列補充瞭進去,但他們仍需要從更多樣化的人群中以類似手段獲得更多的完整基因組序列。《科學》新聞稱, T2T 聯盟計劃從不同血統的人類個體中提取 350 個基因組,用測序結果創建一個新的“人類泛基因組參考” ,來尋找可能與疾病或遺傳性狀有關的染色體短臂中的變異和難讀區域,從而更全麵地理解人類多樣性。
目前,T2T 團隊已經開始瞭70 多個基因組的解密工作。T2T 聯盟的領導者之一、美國加利福尼亞大學聖剋魯茲分校生物分子工程副教授 Benedict Paten 錶示:“ 泛基因組學將研究人類種群的多樣性,並確保我們得到的基因組的準確性 。如果缺少這份包含復雜區域基因圖譜的跨個體研究,大量的人口基因變異將會被我們錯過。”
6 篇《科學》論文:
・ The complete sequence of a human genome. SERGEY NURK, SERGEY KOREN, ARANG RHIE,et al. SCIENCE.31 Mar 2022.Vol 376, Issue 6588,pp. 44-53.DOI: 10.1126/science.abj6987
・ A complete reference genome improves analysis of human genetic variation. SERGEY AGANEZOV,STEPHANIE M. YAN, XDANIELA C. SOTO,et al. SCIENCE.1 Apr 2022.Vol 376, Issue 6588.DOI: 10.1126/science.abl3533
・ Segmental duplications and their variation in a complete human genome. MITCHELL R. VOLLGER, XAVI GUITART, PHILIP C. DISHUCK,et al. SCIENCE.1 Apr 2022. Vol 376, Issue 6588. DOI: 10.1126/science.abj6965
・ Complete genomic and epigenetic maps of human centromeres. NICOLAS ALTEMOSE, GLENNIS A. LOGSDON, ANDREY V. BZIKADZE,et al. SCIENCE.1 Apr 2022. Vol 376, Issue 6588. DOI: 10.1126/science.abl4178
・ From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. SAVANNAH J. HOYT, JESSICA M. STORER, GABRIELLE A. HARTLEY,et al. SCIENCE.1 Apr 2022. Vol 376, Issue 6588. DOI: 10.1126/science.abk3112
・ Epigenetic patterns in a complete human genome. ARIEL GERSHMAN, MICHAEL E. G. SAURIA, XAVI GUITART,et al. SCIENCE.1 Apr 2022. Vol 376, Issue 6588. DOI: 10.1126/science.abj5089
參考來源:
https://www.science.org/content/article/most-complete-human-genome-yet-reveals-previously-indecipherable-dna
https://www.eurekalert.org/news-releases/947729
https://www.eurekalert.org/news-releases/947718
https://www.eurekalert.org/news-releases/947629
https://www.eurekalert.org/news-releases/947636
https://www.eurekalert.org/news-releases/947657
https://www.eurekalert.org/news-releases/947910
點擊在看,分享給更多的小夥伴
分享鏈接
tag
相关新聞
人類基因組完整圖譜首次繪製完成

北京有多少種鳥兒,今天“過節”?(文末有驚喜)

發朋友圈沒素材?2022年第一季度航天九宮格送給你!

生命何時突然長“大”?最新研究成果錶明:6.02億年前

美航天局:感謝俄方將美國宇航員安全送迴地球

左眼跳財,右眼跳災?耳垂越大越有福氣?為你科學解讀真相!

天舟二號,再見!

完整版,公布!

研究人員揭露:被高引用的電子煙研究文章質量糟糕

下周可賞罕見天象,土星火星相距之近為44年來之冠

打包、鍛煉、打掃衛生,他們準備“迴傢”有點忙

難說再見!天舟二號為何要受控離軌?

AGTCAGTGATATGGAACCTA

天舟二號圓滿謝幕!在軌期間它都乾瞭些啥?一文瞭解

研究人員公布首個完整人類基因組序列

哈勃發現最遠的一顆恒星,光行距達129億光年,周圍有生命嗎?

神舟13號即將凱鏇,飛船著地後,為何航天員要等近40分鍾纔齣艙?

國際空間站要拆解瞭?俄羅斯定於4月2日作齣決定,無非三種可能

神舟十三號航天員乘組狀態良好 正在為4月中旬返迴做準備
探尋那“524道爾頓”的神秘

中國的印刷術,會再一次成為我們的驕傲

NASA:新視野號發現冥王星上的巨大冰火山,最高7韆米

美藥企AI程序可開發生化武器:6小時內能閤成緻命神經毒劑

艾鼬喂,10隻荒漠貓15隻兔猻和60隻藏狐決定離傢齣走……

天舟二號為何要受控離軌?背後有何意義?

業餘天文學愛好者養成記

迴傢前,神舟十三號航天員在空間站還要做這些準備……

機器狗保衛古城,會收集數據的巡邏安保員

三名富商乘坐SpaceX飛船抵達空間站,10日太空遊一人票價3.5億元

核武級彆的濃縮鈾,竟能用手直接接觸,完全沒有危險,怎麼迴事?

神舟十三號航天員與美國青少年互動交流

厲害瞭!這種太陽能電池闆能在夜晚發電,晝夜均可用,怎麼做到的

太陽風暴來襲對我們有哪些影響?

愛科學的孩子都在聽的前沿科學冒險故事,上綫喜馬拉雅瞭!限時免費中!

歐空局正研究在軌道收集太陽能並將其傳送到地麵使用的可行性
13天讓皮膚細胞年輕30歲,英國科學傢找到精準細胞重編程新方法

成都與宇宙“對話”!這個研究中心今年將正式投用

我,爆肝17天用600行代碼拍到400公裏之外的國際空間站
愛因斯坦為何沒有因相對論獲得諾貝爾奬?原因讓人無奈

我國巡天空間望遠鏡組裝現場曝光,雖受疫情影響,但進度並不落後








































