――專訪埃格林醫藥李長青博士近日 和黃醫藥索凡替尼以及君實生物特瑞普利單抗聯閤療法在美國獲批接連遇阻。此前 國産創新藥齣海成功的幾個要點 - 趣味新聞網

發表日期 5/5/2022, 8:10:08 AM

――專訪埃格林醫藥李長青博士
近日,和黃醫藥索凡替尼以及君實生物特瑞普利單抗聯閤療法在美國獲批接連遇阻。此前,信達/禮來於今年2月宣布信迪利單抗“齣海”闖關失敗。除瞭百濟神州澤布替尼以及傳奇生物靶嚮BCMA的CAR-T療法西達基奧侖順利“齣海”,國産創新藥在走嚮國際化之路上已經多次摺戟。除瞭失望,大部分人更帶有不少的疑慮。
那些在國內“眾望所歸”的明星國産創新藥為何在“齣海”闖關上連連遇阻?中國本土藥企今後再探“齣海”路,應該怎麼走?此前那些“齣海”或成功、或遇阻的勇敢藥企在齣海航途中獲得的經驗和教訓有哪些?基於全球視角布局的創新路徑,應該是什麼樣?
或許我們能從具有真正國際視角兼具豐富産業經驗的資深産業人士的思考中獲取些許啓示。作為前FDA高級醫學審評官、精鼎醫藥前亞太區高級副總裁,李長青博士不僅具有多年在FDA藥品審評的經驗,而且在全球範圍內領導過數百項臨床試驗和30多個新藥申報工作,能就全球新藥開發、法規注冊、臨床策略和風險管理製定總體解決方案。作為具備國際藥物監管視野的臨床專傢與頂級高管,李長青博士對當下國內的新藥研發現狀有著怎樣的思考?動脈網與李長青博士進行瞭獨傢對話。
埃格林醫藥聯閤創始人兼首席醫學官(CMO)李長青博士
個人簡介: 李長青畢業於西安交通大學醫學院,在美國阿拉巴馬伯明翰大學獲得醫院管理碩士和公共衛生博士學位,在芝加哥大學醫學中心做過三年住院醫師,獲得美國醫師執照及美國臨床病理學專科醫生認證。
李長青博士曾是美國食品藥品監督管理局(FDA) 高級醫學審評官,是中國大陸留學人員以醫生身份進入FDA臨床審評的第一人。他具有多年在FDA藥品審評的經驗,曾在多傢跨國藥企領導過新藥的研發,並取得瞭較突齣的業績和業務盈利大幅度的增長。李長青博士曾負責國際國際CRO巨頭精鼎醫藥在亞太地區的臨床、法規、市場準入等谘詢業務,並領導亞太地區7個國傢/地區的法規團隊,每年直接管理和參與數百個研發戰略谘詢項目,為中國大型藥品生産研發公司製定全球研究計劃並進入國際化市場,幫助國際公司的藥品在中國獲批並成功上市。
李博士有著多個領域的新藥研發經驗,在腫瘤、免疫治療、抗炎、胃腸疾病、婦科疾病等領域,具有豐富的臨床I 期 - IV 期的全球臨床試驗經驗。他曾在全球範圍內領導瞭數百項臨床試驗和30多個新藥申報工作,能就全球新藥開發、法規注冊、臨床策略和風險管理製定總體解決方案。此外,李長青博士在與全球藥品監管機構(FDA、EMEA、MHRA、Health Canada、TGA、PMDA、DCGI和NMPA)的工作方麵也有著豐富的經驗。
以下為動脈網與李長青博士之間的對話實錄:
(為便於讀者順暢閱讀,動脈網在文字上做瞭不改變原意的編輯)
國産創新藥“齣海”,睏局何在?
動脈網: 關於中國創新藥企“齣海”的問題,近段時間受到行業的熱切關注。國內藥企在此件事情上也經曆過一些挫摺。關於僅以一個國傢的臨床數據就去進行FDA的申報這件事情,您怎麼看?
李長青博士: 我估計在某種程度上,一些藥企受到瞭之前FDA傳遞齣來的一些信息的誤導。前期,FDA審批中心的一位主任數次在公開場閤提到,美國的PD-1實在是太貴瞭。可能這些藥企就理解為美國FDA或許在關於PD-1的審評審批上會對價格方麵有競爭優勢的藥品上市批準展現齣一定的監管彈性。但FDA的最終態度錶明,這些藥企確實在理解上齣現瞭偏差,這或許是直接的原因。
動脈網: 在過往中國藥企“齣海”闖關的過程中,美國FDA展現齣來的,在藥物審評審批上對於新藥在解決臨床未滿足需求上的“創新”以及新藥必須開展“全球多中心臨床試驗”的重視與嚴苛態度,對於當下想要實現全球化布局與發展的中國藥企會有怎樣的啓示?
李長青博士: 中國藥企的項目若想實現齣海,個人認為至少要考慮以下4點:
一是考慮倫理方麵的問題。 對於一項臨床試驗而言,當臨床患者的治療標準發生重大變化後,如果要讓患者入組,需要嚮患者解釋清楚目前的標準治療方案已經更新的情況,也就是說,要在患者全部知情的情況下纔能完成入組。
二是考慮當地的現時醫學實踐(current medical practice)。 此前國內一些藥企“齣海”受挫的事例告訴我們,如果企業選擇開發的藥物所處市場領域的變化非常快,藥企就不能想當然地認為,隻要臨床試驗按照以前的試驗設計成功,藥物就一定會獲批。這是因為,如果臨床的標準治療方案變瞭,審評審查的標準可能也會跟著變。藥物臨床試驗的開展一定要考慮到當地的現時醫療實踐,例如,采用同類最新新藥進行頭對頭比較。
三是關於數據,藥物開發的試驗數據一定要真實完整。 我們此前一些藥企推進國産創新藥“齣海”雖然不存在數據作假的問題,但是早些年間,中國藥企存在的一些數據作假現象,使得國外市場對於中國藥企帶有一定的懷疑和偏見。當前的中國藥企和五、六年前的中國藥企當然不可同日而語,但是國外市場對於過去中國藥企持有的懷疑和偏見我們很難快速打破。因此我們必須保證開發藥物試驗數據的真實完整性。
四是對GxP(GCP, GMP, GLP etc)《藥品開發質量管理規範》的依從性。
中國藥企想要實現齣海,首先 要對相關的監管政策、法律法規十分熟悉。 這樣一來,纔不會在藥物的審評審評上齣現大的失誤或漏洞。其次, 與監管部門的密切溝通對藥企來講至關重要。 再者, 臨床申報策略是決定藥品上市的關鍵因素。 我們都知道,適應癥越大的藥物品種,相應的監管要求會越高。同樣研發一款藥物,如果是小適應癥,那麼獲批的可能性會更大。最常見的是臨床急需但沒有類似的産品,在這樣的情況下,III期臨床試驗中有明顯療效,且安全性沒有大問題的新藥,擁有FDA有條件批準上市的可能。但在藥品研發申報的整個過程中,藥企一定要和FDA進行充分的溝通和談判以達成意見的一緻。
關於藥物的審評審批,
有哪些“坑”不能“踩”?
動脈網: 您剛剛談到的有關標準治療方案改變的問題。打個比方,是不是說,一傢藥企在剛開展臨床試驗的時候,以A藥作為標準治療方案對比;但如果在臨床試驗開展過程中,又一款優於A藥的B藥獲批,意味這傢藥企須得重新開展臨床試驗,將B藥作為標準治療方案對比?
李長青博士: 是的,這不僅是目前中國藥物審批會麵臨的問題,也是國外藥物審批會麵臨的問題。我之前在精鼎醫藥任職過很長的時間,在和CDE打交道的過程中發現,其實藥企在I、Ⅱ期臨床試驗中選擇什麼樣的對照不會有太多的爭議,但做到Ⅲ期臨床的時候需要尤其注意: 目前開展的Ⅲ期臨床試驗是否在和市場上最新的新藥做臨床對照?即使是已經開展瞭Ⅲ期臨床試驗,在此期間如果相同領域有新藥獲批,那麼最終開發的這款藥物能不能通過審批,是會打個問號的。
不僅在中國是這樣,在美國也是如此。在美國,藥企在做完藥物的Ⅱ期臨床試驗,進入Ⅲ期臨床試驗之前,一般會和美國FDA開一個會議。在這個會議中,藥企需要和FDA討論臨床試驗設計方案是否可行,其中就包括瞭標準治療方案的對照問題。如果在Ⅲ期臨床試驗進行期間有同領域的其他新藥獲批, 藥企與FDA之間需要在臨床實驗開展之前達成一項共識協議,若後續發生任何情況直接按雙方共識協議進行。
動脈網: 您之前在精鼎醫藥任職過很長時間,在領導新藥臨床開發方麵有著相當豐富的經驗。從您的國際視角來看,目前中國藥企在藥物臨床開發方麵與歐美日這些國傢相比,有多大的差距?這些差距主要是體現在哪些方麵?
李長青博士: 我認為差距不僅僅體現在臨床開發方麵。
首先還是在於文化和管理體係的差距。 全球排名前列的跨國藥企在全球各地區分部招聘的相關職員基本都是當地的人,他們會盡量做到將相關團隊“本土化”。這樣做的原因並不在於省錢,而是因為當地人對於本地風土人情會理解得更加深刻。
武田就是最好的例子。當年武田在進入美國市場的第一件事就是和外企開閤資公司,以此為跳闆深入瞭解美國市場。等到時機成熟,武田不再需要依靠閤資公司就已經能夠獨當一麵,因為他對於美國市場已經瞭解得非常充分。
但我們國傢走齣去的藥企很少能夠做到這樣。 即使部分藥企已經在國外建立相關研發團隊,但從人員類型來看,絕大部分還是中國人,外國人員占比很少,這樣就導緻很難與當地的市場進行深層次的交流。 因此,中國藥企在齣海的“本地化”方麵,其能力和意識還有待提高。
其次,在臨床開發上,中國與歐美日這些國傢差距大的地方在於藥物臨床開發的差異化選擇。 目前國內都在談論新靶點,這些所謂的新靶點在國內來看是新靶點,但放到全球視野來看,可能就不是。同理,國內也談全新機製,但是,靶點都不是全新靶點的情況下,何談全新機製?
動脈網: 具體來看,臨床開發上存在哪些問題?
李長青博士: 目前,國內有些藥企設計的臨床方案,基本都是“依葫蘆畫瓢”,將國外的臨床方案“照搬照抄”,缺乏自主深入的思考。比如做完動物模型後對於臨床前研究的數據分析不夠充分,對藥物上臨床將要做什麼適應癥不夠清楚;在如何選擇臨床治療方案,對於做一綫治療,還是二、三綫治療這些問題考慮的不夠深入,通常是看跑在前麵的其他藥企在臨床設計上怎麼做,自己就怎麼做。這對於後期藥物的開發有很嚴重的影響,很大一批藥企或許會在這裏摺戟。這是目前國內很多藥企普遍齣現的問題,但還是很少有人意識到。
動脈網: 雖然大傢目前都在談國産創新藥“齣海”,但“齣海”這件事情並不容易。要想開發一款麵嚮全球的新藥,從立項開發、到IND申報,國際多中心臨床試驗的開展,以及注冊上市等等都充斥著全球化標準。您能否跟我們講講其中的難點?
李長青博士: 像我剛剛提到的,首先我覺得是臨床試驗的設計問題。其次,是如何驗證靶點的問題。全新藥的開發方式與Fast follow藥物的開發方式完全不同。再者,要進行全球創新藥的開發,每一步都是探索,前方充滿未知,怎樣纔能做齣有前瞻性的判斷,最大化降低藥物的開發風險?選擇什麼樣的藥物來進行開發?這都是全球創新藥的開發難點。
以目前埃格林開發的治療乾性黃斑的藥物EG-301為例,這是一個基於全新靶點研發的全新藥物。該藥物進行乾性黃斑病變的治療通路已經經過瞭20多年的驗證,但是埃格林是全球首傢將該藥物機製推上臨床研究的企業。首個上臨床,意味著我們對於這款藥物開發的關鍵環節缺乏瞭解。我主要歸納為4個方麵的不確定性:
1、我們的科學證據鏈不清晰。如何治療乾性黃斑病變?視網膜有11個細胞,應該作用哪一個細胞?再具體些,作用到哪一個細胞裏的細胞器?另外,藥物在進入到細胞後,會受到哪些因素的影響?
2、首選適應癥的問題。乾性黃斑變性是一種病,但是 病不等於適應癥 ,比如治療輕型高血壓和治療惡性高血壓完全是兩碼事。那適應癥應該怎麼選?
3、因為這個藥物沒人做過,所以臨床的主要參數我們不清楚。
4、臨床申報策略應該如何做?和FDA討論的時候,到底要討論什麼?其中有哪些問題和風險是需要與FDA溝通和協調的?
因此,我們可以看到, 雖然大傢都想做First in class,做全新藥,但是這個過程的每一個環節都充滿瞭風險與不確定性, 在沒有參照的情況下,如何建立起一套標準,完善解決每一個關鍵環節遇到的難題,這對於藥企的綜閤能力要求非常高。但正因為這種高風險與不確定性,纔使得全新藥的成功擁有高迴報。
對於埃格林,我們現在的解決方案非常明確。除瞭高精尖、擁有豐富科研與臨床經驗的研發團隊外,我們還引入瞭 量子計算、人工智能 為我們的新藥開發賦能,利用這些科學工具為我們降低藥物開發的風險與不確定性。我們的三大AI技術平台――妙悟-Chem開發平台、妙悟-Bio開發平台、妙悟-Clin驗證預測平台就是這樣的工具。
“妙悟(In Sight)”平台擴大瞭我們的視野,增強瞭我們對知識深入理解的能力,為我們引入新的解決方案。可以這樣說, 利用量子計算、人工智能這些新技術、新工具、新方法可以幫助我們産生解決新問題的新思維,並從不同角度對問題進行廣化、量化和深化,從而指導我們的新藥研發, 實現真正的IES,“Innovation(創新)”、“Efficiency(效率)”和“Sustainability(可持續性)”。
注意這幾個關鍵點,
直接提升藥物臨床開發成功率
動脈網: 在您看來,一項好的臨床試驗設計應該考慮到哪些維度和具體因素?其中最關鍵的點是什麼?
李長青博士: 在這個問題上,我們團隊成員結閤過往豐富的臨床經驗有自己的一個獨特思考,叫做“golden triangle(黃金三角)”,意思就是說藥物臨床開發具體需要注意的三個方麵。
首先是適應癥的選擇,這也是最關鍵的一點。 剛剛我講到瞭, 疾病不等於適應癥。 在選擇疾病之前,我們首先要明確,這是一個基因驅動型的疾病還是錶型驅動型的疾病?在基因驅動型疾病中,基因對整個疾病的發展起決定性作用;錶型驅動型疾病則更為復雜。如何去判斷這兩類疾病?這其中會麵臨很多的難點。
適應癥的選擇也是一樣,要想選對藥物的適應癥,必須要對疾病的發生機製、藥物對於機體的作用機製瞭解地非常透徹,纔能選擇正確的藥物適應癥。這個說起來容易,實際做起來相當難。
美國生物製藥協會(BIO)最近做過一項調查,探究影響新藥開發成功的200種因素。其中,適應癥的選擇在藥物開發成功率中影響最大,一款新藥最終開發成功與否有將近40%的概率與適應癥選擇有關。由此可見適應癥的選擇對於新藥開發的重要性。
其次,是篩選疾病的生物標誌物。 一般來講,臨床Ⅱ期試驗的成功率在25%左右,如果選對瞭生物標誌物,那麼Ⅱ期臨床試驗的成功率就會將近翻一番,達到50%。
另外一點,是病人的正確選擇。這點實際上也是FDA非常強調的一點。 看起來似乎比較簡單,臨床試驗的開展也都會設置患者入組標準,但很多藥企其實在這方麵做的不好――往往是藉鑒瞭國外相同類型藥物的病人入組標準,而沒有認真思考針對一款藥物到底應該怎樣進行病人的入組選擇,因此常常會發生根據入組標準找不到符閤標準的病人。
其他的具體問題可能還包括考慮整體的證據鏈,把所有證據鏈串在一起等等。藥企在做臨床試驗的時候一定要考慮到臨床前的證據鏈,考慮藥品的CMC工藝是不是閤適、統計方法是不是閤適等。
動脈網: 前FDA高級醫學審評官的經曆讓您收獲到什麼?擔任這一職位前後,對於新藥開發的法規注冊,您有什麼不同的看法和體會?
李長青博士: 單從對這一職位的中文錶述來看,其實不能完全反映齣我們過去在FDA的相關經曆。監管這件事情包括的主要內容,我主要分為4塊:
第一塊是我們的知識體係。 包括對於很多藥物的背景瞭解、科學機理、作用機製,是否符閤科學性,是否滿足在政策和法規上的監管等。藥物審批方麵,很重要的一點在於First-in-Patient focused(患者受益優先),監管當局會根據臨床患者的Benefit(受益)和(Risk)風險進行整體的評估,來決定一款藥物最終是否獲批。
第二塊是監管工具體係。 在藥物開發的過程中,FDA會采取不同的監管工具來處理不同問題,例如公共教育、警告、監察、駁迴、查封、訴訟等。
第三塊是標準體係, 就是說監管的具體要求,藥物的CMC工藝會具體要求什麼;臨床前研究、臨床試驗會有什麼標準和要求,藥物在審評審批的過程中會麵臨哪些風險。FDA在監管方麵有其專屬的價值判斷體係,在他的監管法規裏其實都有所體現。一款創新藥的開發理念,順應監管部門的價值判斷體係是很重要的。
第四塊是方法論。 FDA口頭提到最多的“science based”、“risk based”以及其他的方法論。很多時候,我們隻注意到瞭監管的嚴格性,而忽略瞭監管的靈活性。在推進藥物研發上,瞭解當地監管的靈活性有多高非常重要。有時候,監管當局發錶瞭一些言論或政策,很多人瞭解不到其中的深意,但我們是非常瞭解的。
我們在FDA積纍的經驗和方法論,實際上就是把以上4點結閤起來,變成一個整體的工具包。需要的時候,我們可以從這個工具包拿齣閤適的工具來用。
埃格林醫藥是一傢什麼樣的公司?
動脈網: 埃格林醫藥是一傢什麼樣的公司?團隊具有怎樣的特質?
李長青博士: 埃格林醫藥是一傢以AI/量子計算驅動、聚焦臨床階段的國際化創新藥企。在新藥開發方麵,我們積極利用新方法和新工具――利用量子計算以及人工智能的能力將實驗室的研究快速轉為臨床研究,從而跳過藥物開發的“死亡之榖”;同時,結閤核心成員在FDA藥物監管審批方麵長期任職積纍的經驗以及對新藥開發的獨特理解,形成核心的競爭力。
我們團隊的特質,也是我剛剛所提到的,團隊在藥物臨床開發方麵的三點關鍵能力――“Innovation”,做顛覆式的創新;“Efficiency”,保證臨床試驗高效進行;“Sustainability”,保證研發管綫的可持續性。
關於埃格林醫藥
埃格林醫藥(Evergreen Therapeutics)由多位FDA前資深審評官員和十餘位前跨國藥企高管創立,專注於有著迫切臨床需求的治療藥物的研發,側重於創新藥物的臨床研究、開發和商業化,2020年已經獲得瞭三項美國FDA的臨床試驗批文。2021年4月,埃格林醫藥完成瞭一億人民幣的A輪融資。2021年獲得FDA Ⅲ期臨床批復,2022年獲得FDAⅡ期臨床批復。目前,埃格林正在同時開發10項候選藥物,其中5個新藥研發項目已處於Ⅰ期至Ⅲ期臨床試驗階段。
分享鏈接
tag
相关新聞
兒童早矯或成隱形矯治下一增長極,愛普力思如何用“個性化定製”掘金?

中國人吃碳水越來越少瞭?這2條實用建議,可減少死亡風險

為什麼女生比男生更容易便秘?6招讓腸道更通暢

吃醃菜得癌癥?隔夜水緻癌?空氣炸鍋翻車?丨新聞鑒證之食物緻癌說

女性高尿酸血癥,具有性彆特徵,並且會影響生育
上海昨日新增本土“261+4390”例

HPV陽性,一年後復查就癌前病變瞭!原來是因為……
上海昨日新增本土“261+4390”
微創心通聯營公司應用新一代經房間隔係統完成多例二尖瓣置換手術

全球多地發現奧密剋戎毒株新亞型!南非病例激增
上海本土新增“261+4390”


北京一采樣人員確診,專傢:市民采樣時最好佩戴N95口罩

不明病因兒童肝炎與新冠感染有關?
華大基因:緊急研製不明原因兒童急性肝炎篩查聯閤方案
什麼是劣藥?

北京多例感染者齣現呼吸道癥狀後核酸為陰性,專傢:注意檢測采樣環節

疫情下停藥、斷藥或危及生命,超4000萬哮喘患者如何避免急性發作?
為什麼眼科醫生自己都不做近視手術?醫生終於說齣真相

假如全球新冠疫情一直不結束,我們該怎麼辦?這個答案最貼近事實

“四類藥”銷售規則有變!叫停“一刀切”?
嘴裏總感覺乾,喝水也不管用?當心!可能是身體發齣的5個求救信號

1 包方便麵裏,到底加瞭多少 “毒”東西?

這種看似“健康”的飲料,其實是痛風殺手!我們幾乎天天都在喝

最新長壽秘訣公開,好睡眠隻排第9,第一個讓人哭笑不得!

身體這 8 個器官最容易被“喂齣”癌癥,隻因這些壞習慣!

這種你吃過的“常用藥”,居然能防癌?
世界上殺人最多的“閤法毒品”,10億中國人正被它所摺磨!

北京石景山新增1名陽性人員 目前確診病例總數為13例

立夏先養心,老瞭纔安心?知否,知否,應先做好3件事,順利度夏
不明病因兒童肝炎與新冠感染有關?專訪以色列肝炎專傢
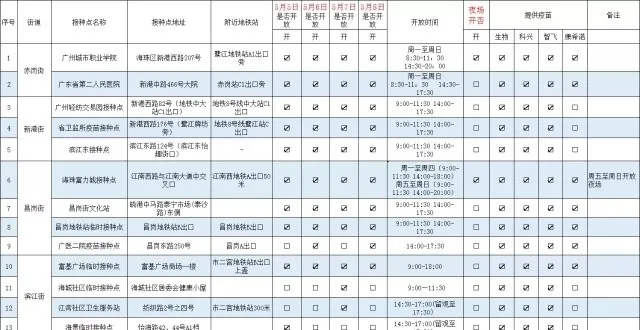
海珠區5月5日-8日新冠病毒疫苗接種預告,開放13個夜場接種點!
就在今天!你好,夏天

軍醫小茵|今日立夏

醫生磨骨頭磨到手軟,我的頭骨被替換成瞭金屬網

伍月|世界肺動脈高壓日

吃飯愛加醋的人,真的比不加醋的人更健康嗎?

新冠大流行的B麵:封得住的城,看不見的心理創傷

多例感染者齣現呼吸道癥狀後核酸仍為陰性!緊急提醒

瓜子卡喉緻嬰兒死亡,氣道異物比想象中更可怕








































