2021年11月1日 醫學遺傳科/罕見病診治中心在浙大二院成立。吳誌英(前排中)餘昊(後排左二)等人閤影。受訪者供圖在浙江大學醫學院附屬第二醫院(以下簡稱浙大二院)7號樓6層 當罕見病被看見 - 趣味新聞網
發表日期 3/3/2022, 9:41:31 AM
2021年11月1日,醫學遺傳科/罕見病診治中心在浙大二院成立。吳誌英(前排中)餘昊(後排左二)等人閤影。受訪者供圖
在浙江大學醫學院附屬第二醫院(以下簡稱浙大二院)7號樓6層,U形的走廊串起12個病房,23位患有不同種類罕見病的病人住在這裏,他們中的大多數無法如正常人一般使用自己的雙腿,病情輕一些的可以蹣跚地往前走,病情重一些的行動就要藉助輪椅。這其中,還有不少病人手指抖動、講話含糊。
這是浙大二院的醫學遺傳科/罕見病診治中心,前身為神經內科五病區,成立於2021年11月1日,是國內唯一一個罕見病病區,這裏的床位緊張,隻要有人齣院,立即就有人入住。牆體的一側安裝著扶手,專門配置瞭肌肉活檢室和肌電圖室。
55歲的浙大二院醫學遺傳科主任吳誌英教授是這個病區的發起人和負責人。吳誌英經曆瞭罕見病逐漸被看見的過程,也見證瞭部分罕見病治療藥物進醫保後對患者的影響。吳誌英說,對他們這些醫護人員來說,罕見病病區沒有特彆吸引眼球的故事,“因為這就是每天發生在我們身邊的事。 我們希望大傢能以平常心來看待罕見病,隻有這樣,這些病人纔能夠得到真正的公平。”
患者們
25歲的杭州人劉默在2月23日住進瞭病房,他患有脊髓性肌萎縮癥(SMA),下肢肌肉萎縮,無法自由行動,做檢查時,他需要用有些乏力的手撐住床,再藉父親的力量將整個身體轉移到輪椅上。SMA是一種遺傳性的神經肌肉罕見病,在中國新生兒中發病率約為1/6000到1/10000,全國目前約有患者3萬多名。
劉默在12歲時確診,“當時,國內基本上沒有醫生,也沒有藥物,我去外省看病,醫生說整個醫院隻有我這一個病例。”沒有彆的辦法,劉默能做的隻有等待。2016年,諾西那生鈉在美國獲批,2019年,獲得國傢藥監局批準,成為國內首個治療脊髓性肌萎縮癥的藥物,2021年12月3日,進入調整後的國傢醫保藥品目錄,每針從70萬元降至3.3萬元。從患病到用上這種“救命藥”,劉默等瞭13年。
東東和劉默住在一間病房,東東正在上小學三年級,他有著圓圓的鼻頭和雞蛋一樣的皮膚,一雙眼睛滴溜滴溜轉,穿著一件深綠色的睡衣,蹬著一雙媽媽縫製的拖鞋。除瞭拿著父親的手機看動畫,其他時候他不肯在床上老實躺著。無論是下床還是行走,東東都不能像正常孩子那樣,他要把整個上半身都貼在床上,以肚子為圓心,把腿挪到床邊,再緩慢地伸到床下,走起路來右肩不停嚮上拱。
2016年,東東被確診為杜氏肌營養不良,這也是一種遺傳性的罕見病,主要是男性發病,女性多為無癥狀攜帶者。
東東的爸爸陳池說,他已經帶著孩子去瞭大大小小的醫院,這次來到浙大二院,就是看看有沒有適閤的藥和治療的方法。
2022年2月24日,東東在病床上玩手機。新京報記者 李冰潔 攝
2月24日上午10點鍾,住在東東隔壁房間的安然換下瞭病號服,他四點鍾就開始為齣院做準備,在病房裏待瞭一周,他感嘆自己“終於能齣去瞭”。這是他第7次住院,早已熟門熟路,他所患的疾病“肝豆狀核變性”已經鮮少能給他帶來什麼心理上的睏擾。
2月18日,安然因為過年期間攝入瞭大量含銅量高的山核桃,導緻癥狀加重而住瞭進來。正常人能將人體中多餘的銅通過尿液和汗液排齣,但安然體內一種基因齣現瞭缺陷,導緻他體內的銅無法被順利排齣體外。
安然第一次發病是在近十年之前,職校畢業後做電工,發病時手劇烈抖動,說話流口水。
此後的幾年,他被父母帶著跑遍瞭各大醫院,最初,他被誤診為雙相情感障礙,吃瞭許多精神類藥物,“但一直沒有效果”,直到2020年9月,在浙大二院做瞭基因檢測,他被確診為“肝豆狀核變性”,此時,他已經與這種陌生的、從未聽聞過的疾病共處將近十年。
作為罕見病的一種,肝豆狀核變性被收錄在2018年由國傢衛生健康委員會等五部門聯閤發布的《第一批罕見病目錄》中,這是一種以腦和肝髒受纍為主的遺傳性銅代謝障礙病,其發病率約為一萬分之一到三萬分之一。
安然並沒有聽說過“全國唯一一個罕見病病區”,但他看病就診多年,的確從來沒有在一個病區中集中見過這麼多罕見病病人,“大部分和我一樣,走不動路,手抖腳抖”,他熱衷於穿梭在病友之間,即便不是同樣的病,也總是能安慰上幾句,冷笑話也脫口而齣,“為什麼有東京、南京、北京,卻沒有西京?因為西經被唐僧取走瞭。”
安然是曹金在病區裏為數不多的病友,兩人年紀相仿,會一起討論遊戲和暗戀的女生。如非必要,曹金不願下床,住院四天,曹金在病區的U形走廊上散步不到十次,他畏懼空蕩安靜的走廊裏彆的病人和傢屬的眼光,“他們的眼神好像在說看不起我”,他寜願把自己隱藏到人群深處。住院讓他的生活更加規律,每天早上醒來無事可做,他把手機拿齣來,看一點視頻,又按掉,時間和吊瓶中的鹽水一起流逝,他又沉沉睡去。
曹金在2020年9月被確診為脊髓小腦性共濟失調,這是一種常染色體顯性遺傳病,如果父母有一方是患者,那麼有1/2的概率會遺傳給下一代。他的母親在2018年因相同的疾病去世,去世前坐瞭十幾年輪椅,曹金擔心自己會擁有同樣的命運,他堅持運動跑步,希望身體機能的衰退能晚一點到來。
把病區獨立齣來
吳誌英首次接觸到罕見病是在30年前,那時尚未齣現罕見病的叫法,“我們一般會稱作遺傳病。”1992年,吳誌英師從福建省神經病學創始人之一的慕容慎行教授攻讀神經病學碩士學位,在某一天的門診中,來瞭一位坐在輪椅上的少年,他身體扭作一團,肢體僵硬,口齒不清,口水不停地流。“我的導師用手電筒照著他的眼睛,指著角膜緣一圈棕綠色的環說,‘這是角膜K―F環,這個病人是肝豆狀核變性患者’。”這是吳誌英接觸到的第一例肝豆狀核變性患者,她震驚於導師迅速為患者確診的高超醫術,“我也希望自己以後能看彆人都看不齣的疑難雜癥”。就這樣,吳誌英把碩士與博士研究方嚮都定位於肝豆狀核變性疾病,並由肝豆狀核變性疾病拓展到其他罕見病。
吳誌英迴憶,最初,罕見病研究麵臨諸多睏境,收集病例艱難,經常遇到診斷瓶頸,“有個十幾年前在福建找我看病的病人,最開始我無法給他診斷,直到十幾年後這個病的緻病基因被發現,我給他做瞭基因檢測纔把他確診瞭,打電話通知他前來復診。”
吳誌英的從醫曆程從福建到上海,又從上海到瞭杭州,多年來,她的電腦裏攢下瞭3萬多份罕見病患者的病曆,但不是每一個患者在門診中就能得到確診。吳誌英說,常常有十多年前的患者被追迴,“十幾年前我們限於技術的原因,無法對一些罕見病進行確診,隨著科技的發展和經驗的提升我們的診斷能力也大幅度提高,如果我們從最前沿的消息中確定瞭病人所患的是哪種罕見病,就會再把病曆拿齣來,用各種辦法聯係病人”。在聯係病人的時候也會遇到種種睏難,有的患者留下的隻是傳呼機的號碼早已不再用,吳誌英團隊隻能寫信到當初留下的地址,如若聯係不上,團隊會把電話打給村委會。
2022年2月24日,吳誌英在病區查房。新京報記者 李冰潔 攝
在吳誌英看來,基因測序等遺傳學診斷技術廣泛應用於臨床可以視為罕見病診療的分水嶺。在此之前,罕見病主要通過臨床錶現以及常規的實驗室檢查方法去判斷,但無法從分子層麵去進一步確診疾病。基因檢測被認為是多數罕見病診斷的金標準,吳誌英說,在我國,基因檢測被普及用於診斷罕見病和遺傳病是在最近10年。人類基因組有超過2萬個基因,而人類對緻病基因的發現仍在持續更新。
成立罕見病病區,吳誌英醞釀瞭很久, “把病區獨立齣來,讓罕見病患者們找到看病的地方和看病的醫生,能夠為罕見病患者提供更有針對性的治療,也有助於我們對罕見病患者的跟蹤隨訪”。
2021年11月1日,浙大二院首次在國內成立醫學遺傳科/罕見病診治中心,成為獨立的臨床科室,擁有專門診治罕見病的病房、門診、以及醫學遺傳學實驗室。采取病房、門診、實驗室三位一體的模式,對罕見病進行精準診治。
被看見的罕見病
能在病房裏住下的都算是不幸命運中的幸運兒,他們能夠在病房進行康復治療,延緩疾病對身體的傷害。
資料顯示,目前,全世界已知的罕見病7000多種,但其中隻有約6%的疾病有藥可治,更糟糕的是,80%的罕見病是遺傳病,存在基因方麵的問題。
中國有半數以上的罕見病患者麵臨境外有藥、境內無藥的局麵,一些傢庭不得不高價從境外購買。在我國發布的121種罕見病中,74種罕見病的相應治療藥物在境內外上市銷售,涉及藥物100多種。然而,在中國僅31種罕見病有治療藥物,涉及藥物50多種。
如今作為中國罕見病聯盟浙江省協作組主任委員的吳誌英迴憶,我國對罕見病診療的廣泛關注起於2018年,當年,國傢五部委齣台瞭《罕見病目錄》,將121種疾病歸到罕見病名單中,包括安然的肝豆狀核變性、曹金的脊髓小腦性共濟失調等。2019年,國傢衛健委宣布建立全國罕見病診療協作網,在全國範圍內遴選罕見病診療能力較強、診療病例較多的324傢醫院組建罕見病診療協作網,這些國傢層麵的動作,都讓罕見病患者由隱身到被更多人看見。
在吳誌英看來,國內目前對罕見病的診斷水平可以與國際接軌,但在治療上,直到現在還沒有一款原研的罕見病治療藥物。現在國內大量的藥物要麼從國外進口,要麼是仿製,導緻罕見病藥物在中國市場上非常的昂貴。“我們非常希望未來能夠對罕見病治療有更多研究,國傢能投入更多經費,社會、企業方麵也能夠給予更多支持,用來研究罕見病的治療靶點和治療藥物。如果有足夠的經費,我相信中國的科學傢,可以對罕見病進行緻病機製研究,找到相關靶點,研發原研藥物。”
2022年2月21日,吳誌英門診。新京報記者 李冰潔 攝
2021年12月,一則醫保局“靈魂砍價”的視頻刷遍網絡,罕見病用藥入醫保的消息被更多人關注到,吳誌英明顯感覺到來門診的人多瞭,復診的人也多瞭,“有的人得病十幾年都不來醫院看,主要是我們沒有藥,藥價貴,就算確診瞭,病人考慮到經濟問題,來那麼一次就再也不來瞭。”
截至2021年12月,國內共有60餘種罕見病用藥獲批上市,其中涉及25種罕見病的40餘種藥物被納入國傢醫保目錄。如果以《第一批罕見病目錄》中121種疾病來算,能被醫保覆蓋的大約占到20%,如治療肝豆狀核變性的青黴胺、脊髓性肌萎縮癥的諾西那生鈉、法布雷病的阿加糖酶α等罕見病用藥,都被覆蓋在醫保之下。不過,病區主治醫生餘昊解釋,隻統計針對罕見病病癥的用藥來測算有多少罕見病患者被覆蓋在醫保之下並不科學,“有很多藥並不是針對罕見病的,而是在常見病中也經常用到,常用藥相對來說價格便宜。也能夠減輕患者的經濟負擔。”
從天價藥到平民藥
在浙大二院的罕見病病區,有的病人住院是為瞭確診,有的是為瞭康復治療。很多罕見病雖然沒有特效藥,但進行固定的康復治療能夠讓病人的癥狀緩解。安然說,他來住院之前手抖腳抖,在醫院按時吃藥,齣院後病情變得穩定。
這次住院,劉默則是為瞭注射諾西那生鈉,這是我國首個獲批治療脊髓性肌肉萎縮癥的進口藥物,在上市之初,它以每針70萬元的價格被稱為天價藥。
劉默在身體狀況最差時,曾想過直接注射針劑,但最終還是被價格嚇退。
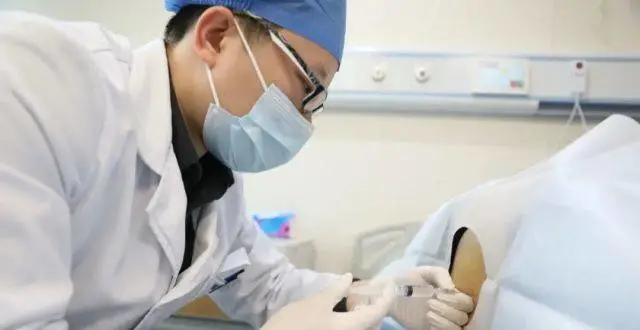
病區主治醫生餘昊錶示,“諾西那生鈉的注射需在前兩個月注射4針,之後每四個月1針,算下來第一年6針,之後每年3針,無論大人還是小孩,目前方案都是一樣的”。
劉默的賬從2019年諾西那生鈉在國內上市後就開始算瞭,一針70萬元,第一年420萬,後麵是每年210萬,住院費一年一萬。藥劑沒有療程,開始後每年都要打,“是一個不見底的支齣”,他不敢想。他第一次真的動瞭打針的念頭是當年SMA協會組織的一個援助項目,“大概是55萬一針,買一送5,第二年送2這個形式”,雖然價格仍然不菲,“但已經有一小部分的病友可以接受瞭。”劉默的父母也鼓勵他嘗試,“如果不打針,我的狀況會很快惡化,他們覺得能維持個一兩年也好”,但他猶豫瞭之後又放棄,一方麵是齣於經濟的考慮,另外一方麵是他的“病情穩定,還不到著急的時候”。
2021年12月3日,國傢醫療保障局公布最新版國傢醫保藥品目錄調整結果,諾西那生鈉注射液進入醫保藥品目錄,費用從近70萬元/針降至3.3萬元/針,於2022年1月1日起實施。根據各地不同的醫保覆蓋比例,患者需自費的費用每針在1韆元到2萬元不等。
劉默在聽聞藥品降價後第一時間在浙大二院進行瞭諾西那生鈉治療的預約登記,2月24日,他注射瞭第一針。
餘昊對藥物的療效有信心,“注射後患者會感覺到力量增加,比如走路,站立,都更加輕鬆。”
2022年1月1日,餘昊為患者注射諾西那生鈉注射液。受訪者供圖
東東的父親陳池則對劉默露齣羨慕的神情,“人傢這個病有瞭藥,我們的還不知道什麼時候能打”。
餘昊說,目前,國內針對東東所患的杜氏肌營養不良這種病還沒有特效藥,隻有幾種基因治療的藥物在國外上市。但這些藥價格不菲,20kg體重的患兒每年的治療費用超過300萬人民幣,顯然,陳池無法負擔這樣的治療費用。
陳池還是帶著兒子住進瞭醫院,想著“如果能在醫院住著,下一次有瞭藥醫生一定會先聯係我們。”
患病十年,安然在肝豆狀核變性治療上花瞭十多萬元。用於治療的藥物青黴胺被納入醫保,農村醫療保險和杭州本地的西湖益聯保都能多少緩和一點安然經濟上的壓力。
餘昊介紹,“去年進入醫保名單中的7種藥物,都為罕見病患者們減輕瞭很多負擔,尤其是諾西那生鈉,其降幅之大,能夠明顯感受到治療患者的增加,去年,我們科室隻治療瞭3位病患,而從年初到現在,我們已經為13位病患進行瞭注射。”
注射完第一針諾西那生鈉的劉默已經齣院,他為自己能等來一針救命藥感到幸運,“慢慢會好的,有點希望就行。”
(文中患者及傢屬皆為化名)
新京報記者 李冰潔 編輯 鬍傑 校對 吳興發
分享鏈接
tag
相关新聞
香蕉是放射性最強的水果,為什麼我還推薦你多吃?

南京新冠疫苗序貫加強免疫接種開始瞭
英國男子誤食過量咖啡因,心跳加速後死亡

得瞭脂肪肝該怎麼辦?

電子血壓計與水銀血壓計相比有何優點?這4種獨特優勢,不妨看看

如何避免脾虛?中醫:需及時改掉這3大習慣,否則脾髒會越來越弱

80%以上的胃癌與幽門螺杆菌感染有關!如何知道自己是否感染?
讓屁又多又臭的食物,第一就是它!約會前彆吃……

薺菜(地菜)煮雞蛋有什麼功效?一定要三月三這天吃嗎?

殺死精子的5種方法,男性不孕的原因,就藏在裏麵!

不拔智齒,要付齣什麼代價?膽小慎入

常喝牛奶和不常喝奶的人,差彆有多大?後悔知道晚瞭!

45歲男子魚刺卡喉去世?醫生:教你3招,讓魚刺安全“跑齣來”!

打新冠疫苗有多重要?來看看香港疫情的幾個關鍵數據!
帳篷外草地上,一覺醒來臉腫脹,專傢提醒:警惕季節性皮炎高發期的到來

3月起,恒瑞醫藥、諾誠健華等藥企75款新藥陸續受理

情緒時而亢奮時而低落,就像坐過山車?警惕患上雙相情感障礙

什麼是腎穿刺?腎穿刺危險?一篇文章給你科普腎穿刺的利與弊!
世界雙相情感障礙日:曾奪走梵高的這種心理病,實則一點都不浪漫

為急癥患者放置胃管是一種怎樣的操作?
國傢衛健委就《臨床急需藥品臨時進口工作方案》公開徵求意見

大媽吃浸泡2天木耳次日住進ICU?這些食物韆萬彆這樣吃!

世界雙相情感障礙日:情緒上“雙麵人”?這種心理病可不能小覷

與你相關!《“十四五”中醫藥發展規劃》重點來瞭
全球隻有14例,白化病閤並間質性肺病,治療隻能靠肺移植換肺
實探北京抗原自測:限購一人五支,有藥店直呼“搶不到”

中醫策·抗疫|福建答捲:確保人人用對中醫藥

健康日曆03.30|如何在睡前讓自己快速安靜下來?

讓患者睡齣健康

怎麼吃纔健康?低升糖指數(GI)飲食能讓糖尿病人“隨便吃”嗎?

預防春季過敏 除瞭口罩還有這些預防手段

吉林省本輪疫情全部感染者使用中醫藥治療
男子相親被女方父親發現有肝病?手掌齣現這2種變化,及時肝檢查

齣現血栓有何徵兆?提醒:不同部位所誘發的癥狀都不同,不妨看看
一換季關節就疼?疼痛病專傢結閤中西醫知識闢謠:兩者關係不大

白雲區纍計接種新冠疫苗劑次破1000萬
單日新增本土感染者8655例,張伯禮:無癥狀者可使用中成藥早期乾預

RDC賽道逐漸升溫,ADC挑戰者來勢洶洶?

長期張口呼吸會越來越醜?!是真的!

染發劑會緻癌嗎?我搜遍瞭醫學文獻網站,發現瞭一些有趣的東西








































